
US District Court, meanwhile, orders all sealed documents in Propecia litigation to be unsealed
February 1, 2021
Dear Friends:
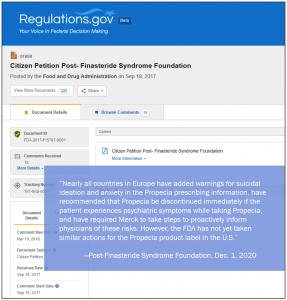 The PFS Foundation has filed supplements to its FDA Citizen Petition requesting that the agency “immediately require withdrawal of marketing approval for Propecia…because the risk of serious injury from the drug outweighs its limited benefits.”
The PFS Foundation has filed supplements to its FDA Citizen Petition requesting that the agency “immediately require withdrawal of marketing approval for Propecia…because the risk of serious injury from the drug outweighs its limited benefits.”
The two supplements were filed Dec. 1, 2020 and posted publicly late last month on Regulations.gov, a US Federal government website that serves as a document repository allowing citizens to participate in the rulemaking processes of some government agencies.
Petition Supplement 1 contains scientific research, epidemiological data and other pertinent information that was published after our Citizen Petition was filed on Sept. 19, 2017, including:
(a) Animal Studies
- Treatment of male rats with finasteride, an inhibitor of 5alpha-reductase enzyme, induces long-lasting effects on depressive-like behavior, hippocampal neurogenesis, neuroinflammation and gut microbiota composition
- The Steroidogenesis Inhibitor Finasteride Reduces the Response to Both Stressful and Rewarding Stimuli.
(b) Clinical Studies
- Altered methylation pattern of the SRD5A2 gene in the cerebrospinal fluid of post-finasteride patients: a pilot study
- Penile vascular abnormalities in young men with persistent side effects after finasteride use for the treatment of androgenic alopecia, Finasteride and Suicide: A Postmarketing Case Series
- Investigation of Suicidality and Psychological Adverse Events in Patients Treated with Finasteride.
(c) Drug Regulatory Agencies Outside the US. The foundation writes:
Nearly all countries in Europe have added warnings for suicidal ideation and anxiety in the Propecia prescribing information, have recommended that Propecia be discontinued immediately if the patient experiences psychiatric symptoms while taking Propecia, and have required Merck to take steps to proactively inform physicians of these risks. However, the FDA has not yet taken similar actions for the Propecia product label in the United States:
- Health Canada: Summary Safety Review for Propecia and Proscar
- Medicines and Healthcare Products Regulatory Agency (UK): Propecia Summary of Product Characteristics
- European Medicines Agency: CMDh position regarding anxiety warning on Propecia Summary of Product Characteristics, EMA
- Federal Institute for Drugs and Medical Devices (BfArM Germany): Finasteride Red Hand Letter
- National Agency for the Safety of Medicines and Health Products (ANSM France)
Petition Supplement 2 refers exclusively to a Reuters report headlined Court let Merck hide secrets about a popular drug’s risks.
Published Sept. 11, 2019 after a yearlong investigation, the story by Dan Levine uncovered testimony by former Merck executives in the US Propecia litigation suggesting that the pharmaceutical giant downplayed the drug’s side effects during clinical trials. Specifically, Merck found evidence of persistent side effects in their original clinical trials for finasteride but failed to disclose such in their warning label.
Compounding this lack of transparency, the judge in the litigation, Brian Cogan, inexplicably allowed Merck and plaintiffs’ lawyers to keep information submitted in court confidential. As Levine, whose report also broke the news of our Citizen Petition, explained:
Some of these documents slipped through cracks in the wall of secrecy. One was inadvertently entered into the public record, staying in the open for a year before being sealed, but in the meantime, it made its way into an obscure public filing, where Reuters found it. The other was faultily redacted, making it possible for this reporter to read it.
Among those documents were depositions of Charlotte Merritt, who oversaw regulatory activity for Propecia, and Paul Howes, who headed up marketing for the prescription medication.
 Merritt admitted that, in 2002, four years after Propecia came to market, Merck altered the drug’s label for sexual adverse events, from “resolution occurred in all men who discontinued therapy with Propecia” to “resolution occurred in men who discontinued therapy with Propecia.” Then she testified that Merck deleted the word “all” because of evidence from the clinical trials that adverse events did not clear up after patients quit the drug.
Merritt admitted that, in 2002, four years after Propecia came to market, Merck altered the drug’s label for sexual adverse events, from “resolution occurred in all men who discontinued therapy with Propecia” to “resolution occurred in men who discontinued therapy with Propecia.” Then she testified that Merck deleted the word “all” because of evidence from the clinical trials that adverse events did not clear up after patients quit the drug.
Howes admitted that Merck knew that warnings of sexual side effects, especially persistent to permanent side effects, would have negatively impacted Propecia sales.
A day after Levine’s story broke, Reuters filed a motion in US Federal Court to unseal all documents filed in the Propecia litigation. The largest news agency in the Western world argued that “This is a case of tremendous importance that has been sealed without on-the-record findings explaining that sealing. The First Amendment precludes such an outcome.”
Sixteen months later, on January 25, US Magistrate Judge Peggy Kuo approved Reuters’ motion, clearing the way to make public all the Merck documents in Propecia-related litigation.
In her decision, Kuo noted that Merck’s arguments for keeping the lid on those documents “are so weak that they would not overcome even a low presumption of access under the common law.”
 Anyone living in the US who suffers from PFS should report his/her symptoms to the US Food and Drug Administration. Anyone living outside the US who suffers from PFS should report his/her symptoms to the US Food and Drug Administration as well as to his/her national drug-regulatory agency, as directed on our Report Your Side Effects page.
Anyone living in the US who suffers from PFS should report his/her symptoms to the US Food and Drug Administration. Anyone living outside the US who suffers from PFS should report his/her symptoms to the US Food and Drug Administration as well as to his/her national drug-regulatory agency, as directed on our Report Your Side Effects page.
If you or a loved one are suffering from PFS, and feeling depressed or unstable, do not hesitate to contact the PFS Foundation via our Patient Support hotline: social@pfsfoundation.org. Thank you.
Related News
Regulatory Update: France’s FDA-Equivalent Agency Reissues Finasteride Warning (Feb. 2, 2019)
Regulatory Update: MHRA Issues Drug Safety Update on Finasteride (May 26, 2017)